Every year, thousands of patients board international flights to receive medical treatments their doctors back home cannot legally offer. These are not desperate people chasing miracle cures on the internet. Many of them are well-researched, well-insured patients who have worked through the conventional medical system and found it lacking. The treatment they are seeking is not unproven. The science behind it is published in peer-reviewed journals. Clinical trials are actively studying it at major university hospitals in the United States. And yet, a physician in Chicago or Houston cannot administer it without risking federal prosecution.
The reason comes down to regulation — specifically, the fundamental differences between how the United States and Colombia classify, oversee, and permit stem cell-based treatments. Understanding those differences matters whether you are a patient weighing your options, a caregiver doing research on behalf of a loved one, or simply someone trying to make sense of why medical access varies so dramatically depending on where you happen to live.
This article is not an argument that all stem cell treatments are safe or that regulatory oversight is unnecessary. It is an attempt to explain, accurately and honestly, why the legal divide exists, what it actually reflects, and what it means for patients who are considering treatment outside the United States.
How the US and Colombia Classify Stem Cell Treatments Differently
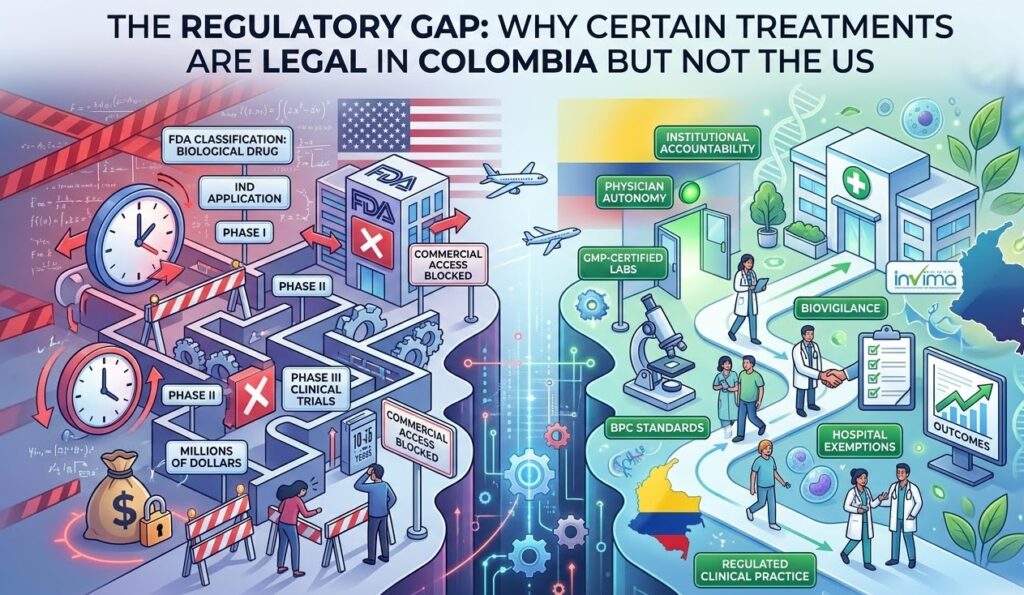
The starting point for understanding the regulatory gap is classification. In the United States, the Food and Drug Administration (FDA) classifies most stem cell treatments as biological drugs. That classification is not incidental. It determines everything: the legal pathway a treatment must follow, the evidence required before it can be offered to patients, and the consequences for practitioners who administer it without that authorization.
Under 21 CFR Part 1271, the FDA applies two key standards to determine whether a cell-based product requires full drug approval. The first is whether the cells have undergone “more than minimal manipulation” during processing. The second is whether the treatment is being used for a “homologous use,” meaning the cells are performing the same function in the recipient that they performed in the donor. If a treatment fails either standard, the FDA treats it as a new biological drug, which means it must go through an Investigational New Drug (IND) application, followed by Phase I, II, and III clinical trials, before it can be offered commercially.
In practice, this framework blocks the vast majority of stem cell treatments from reaching patients outside of clinical trial settings. The exceptions are narrow. Bone marrow transplants for blood cancers have a long track record and regulatory recognition. A small number of CAR-T cell therapies have received full FDA approval for specific blood cancers. Beyond those, most cell-based therapies remain in a legal gray zone where offering them to a paying patient, even with full informed consent, exposes a physician to serious regulatory and criminal liability.
Colombia operates under a different system. INVIMA, the Instituto Nacional de Vigilancia de Medicamentos y Alimentos, functions as Colombia’s equivalent of the FDA. But INVIMA’s framework distinguishes between several categories of treatment delivery that the FDA largely collapses into one. Colombia recognizes formal research protocols, hospital-based exemptions for compassionate or advanced care, and regulated clinical practice within accredited institutions. These categories create legal pathways that simply do not exist in the same form under US law.
The Colombian framework is grounded in Resolution 2378, which establishes Good Clinical Practice (BPC) standards, along with a GMP (Good Manufacturing Practice) certification system for laboratories that process biological materials. A clinic or hospital that operates a GMP-certified lab, follows INVIMA’s biovigilance requirements, and administers treatments within approved institutional protocols can legally offer certain autologous stem cell therapies, meaning therapies using a patient’s own cells, without those cells being classified as a new pharmaceutical drug requiring full market authorization.
That is the core of the regulatory gap. It is not that Colombia has no rules. It is that the rules are structured around institutional accountability, physician oversight, and manufacturing standards rather than requiring every individual treatment to go through a decade-long pharmaceutical approval process.

Why the FDA’s Framework Is So Restrictive
To understand the FDA’s current posture, you have to go back to 1961. That year, a sedative called thalidomide was being prescribed widely in Europe and other parts of the world to help pregnant women manage nausea. It caused severe birth defects in thousands of children. The United States largely avoided the disaster because an FDA medical officer named Frances Kelsey refused to approve the drug without more safety data. The near-miss accelerated the passage of the Kefauver-Harris Amendment in 1962, which fundamentally changed the standards for drug approval in the US by requiring manufacturers to prove both safety and efficacy before bringing a product to market.
That amendment set the institutional DNA of the FDA for the next six decades. The agency’s culture became defined by a particular kind of caution: the political and ethical cost of approving something that later causes harm is far greater, from a regulatory standpoint, than the cost of keeping something off the market that might have helped people. That asymmetry shapes every major decision the FDA makes.
When biological therapies began developing in the late 20th century, the FDA extended its drug approval model to cover them, eventually creating the biologics framework that now governs cell therapies. This made sense for certain products, particularly drugs derived from biological sources that are manufactured at industrial scale and administered uniformly to large patient populations. But it created serious problems for personalized cell therapies, where each treatment is derived from and tailored to a specific patient.
The cost of bringing a new biological drug through the FDA’s full approval pipeline now runs somewhere between one billion and two billion dollars, and the timeline typically stretches between ten and fifteen years. That pipeline was designed for pharmaceutical companies with blockbuster-drug economics. It was not designed for a therapy where a clinic processes a patient’s own fat tissue or bone marrow, concentrates the stem cells, and reintroduces them into the same patient’s body in a clinical setting.
The mismatch is structural. The FDA’s regulations do not have a well-developed middle category for personalized, practitioner-administered biological therapies that are not being manufactured and sold as a commercial product. Attempts to create more flexible pathways, including the Regenerative Medicine Advanced Therapy (RMAT) designation introduced in 2016, have helped move some treatments through trials faster, but they have not changed the fundamental requirement that treatments must complete the drug approval process before being offered outside a clinical trial.
Industry dynamics also play a role. Pharmaceutical and biotech companies that have invested billions in their own cell therapy pipelines have little incentive to advocate for regulatory frameworks that would make it easier for smaller clinics to offer competing treatments. The existing system, whatever its scientific justifications, also functions as a barrier to entry that protects established players.

Why Colombia’s Framework Allows More
Colombia’s regulatory philosophy is not permissive in the sense of being careless. It is permissive in the sense of being structured around different assumptions about where the appropriate locus of oversight should be.
The FDA’s approach essentially says: prove the treatment is safe and effective at the population level before any individual patient can receive it. Colombia’s INVIMA approach says: ensure that the institution administering the treatment meets rigorous standards for manufacturing quality, clinical practice, and patient monitoring, and then allow trained physicians to apply their professional judgment within that framework.
This reflects a broader tradition in Colombian and Latin American medicine of giving significant weight to physician autonomy and institutional accountability. A treatment does not need to be universally approved at the national regulatory level if it is being administered by credentialed specialists in a facility that meets documented quality standards and maintains active biovigilance.
The GMP certification system is central to this. In Colombia, a stem cell laboratory seeking to operate legally must meet GMP standards that govern everything from the physical environment of the lab to the qualifications of the technicians, the documentation of every processing step, and the chain of custody for biological materials. These are not trivial requirements. A GMP-certified facility in Colombia operates under standards that are broadly comparable to those required of biological product manufacturers in Europe and the United States.
On top of GMP certification, INVIMA requires ongoing adverse event reporting and biovigilance. Clinics must track patient outcomes and report complications. This creates a real-time safety monitoring system that informs regulatory decisions over time. It is outcomes-based oversight rather than pre-market gatekeeping.
Colombia has also made a deliberate national investment in medical tourism as an economic sector. The country has developed significant healthcare infrastructure, and its physicians train in programs that meet international accreditation standards. The regulatory environment for advanced therapies is part of a broader positioning effort. Colombia wants to attract patients who cannot access or afford certain treatments in their home countries, and the legal framework for stem cell treatment has developed in ways that support that goal without sacrificing the institutional standards that make the treatments legitimate.

The Safety Question
Any honest discussion of the regulatory gap has to address the safety question directly, because it is the most important one.
The FDA’s supporters would argue that the agency’s restrictive framework exists for good reasons, and they are not entirely wrong. Stem cell-based treatments vary widely in quality. There are clinics around the world, including in the United States before recent enforcement actions, that have administered poorly processed cells in unhygienic conditions with no meaningful follow-up care, causing serious harm to patients. The FDA’s 2017 enforcement crackdown on unlicensed US stem cell clinics was, in part, a response to documented patient injuries.
Those cases are real, and patients should take them seriously. But they do not describe what happens at a GMP-certified, INVIMA-registered clinic operating under documented clinical protocols. The risks associated with rogue operators are not the same as the risks associated with regulated clinical practice.
It is also worth being honest about what FDA approval actually means. FDA approval means that a specific product has cleared a specific regulatory process. It does not mean that approved treatments are universally the safest available, or that unapproved treatments are universally dangerous. Many treatments that are standard of care in Europe and Asia have not been FDA-approved because no company has gone through the expense of the US approval process, not because the evidence is lacking.
Several of the stem cell protocols most commonly administered in Colombia are being actively studied in registered clinical trials at American universities. The scientific community is not in dispute about whether the underlying biology is plausible. The dispute is regulatory and economic, not scientific.
Japan, Germany, and Panama, among others, have developed regulatory frameworks that allow cell-based therapies to reach patients under physician oversight without requiring full pharmaceutical-style approval. Colombia is part of a broader international trend, not an outlier. A patient receiving stem cell therapy in Colombia at a credentialed facility is operating within a regulated system that has parallels in multiple other developed countries.
The meaningful safety distinction is not between the United States and Colombia. It is between reputable, credentialed providers that operate under GMP certification and INVIMA oversight, and unregulated operators that offer treatments with no institutional accountability. That distinction exists regardless of national borders.
What This Means for Patients Considering Treatment in Colombia
For a patient weighing whether to seek treatment in Colombia, the regulatory context matters, but it should not be the only factor. The practical questions are more specific.
The baseline requirements for any provider worth considering are GMP certification for the laboratory and active INVIMA registration for the institution. These are not marketing claims. They are verifiable. A reputable clinic will provide documentation of both without being asked. If a clinic is vague about its regulatory status or cannot produce lab certification records, that is a serious warning sign regardless of how the website looks or how the staff presents themselves.
Beyond credentials, patients should ask specific questions about the treatment protocol: where the cells come from, how they are processed, what concentration and viability standards the lab uses, how the treatment is administered, and what follow-up monitoring is included. Good providers will have clear, documented answers to all of these questions. They will also conduct a clinical evaluation before accepting a patient, because not everyone is a candidate for every protocol.
Guaranteed outcomes are a red flag. No honest provider guarantees results from a biological therapy, because individual response varies based on factors including patient age, baseline health, the specific condition being treated, and timing of treatment relative to disease progression. A provider that promises specific outcomes is telling you more about their sales approach than their medicine.
Patients should also plan for continuity of care. Bring complete medical records to Colombia. Get full documentation of the treatment protocol and any cells processed, and bring that documentation home. Find a physician in your home country who is willing to review what was done and provide follow-up care. Some patients encounter resistance from domestic physicians who are skeptical of treatments administered outside the conventional approval process, but that is a conversation worth having directly rather than avoiding.
The conditions with the strongest published evidence base for stem cell intervention include certain orthopedic applications such as osteoarthritis of the knee and hip, some autoimmune conditions, and certain neurological applications where conventional treatment options are limited. The evidence varies by condition and by protocol, and condition-specific content on this site goes into more detail on individual applications.
The Future of Stem Cell Regulation
The FDA is not static. The RMAT designation, created under the 21st Century Cures Act, gives certain regenerative medicine products an accelerated development pathway that includes more frequent interaction with the FDA, rolling review, and priority review designation. A small number of cell therapies have received RMAT designation and moved through trials faster as a result.
But RMAT does not change the fundamental requirement of completing clinical trials before a treatment can be offered commercially. The program accelerates the process; it does not create a physician-practice exemption or a hospital exemption pathway like those that exist in Colombia. For most patients who need treatment now, the RMAT program is years away from being relevant to their situation.
The more significant long-term question is whether the US will move toward an outcomes-based or risk-stratified regulatory model that creates space for personalized cell therapies to reach patients under physician oversight without requiring full pharmaceutical-scale approval. There is active academic and policy debate about this, and some members of Congress have advocated for expanded right-to-try frameworks that would apply to regenerative therapies. Whether that advocacy produces meaningful legislative change remains to be seen.
Colombia’s regulatory framework is also evolving. INVIMA has been on a sustained modernization path, aligning its standards more closely with international benchmarks, and there is institutional momentum toward making Colombia a recognized center for regenerative medicine research as well as treatment. The institutions operating in this space, including those that have invested in GMP infrastructure and clinical research programs, are part of that process.
The broader international trend points toward a bifurcated future. Countries that develop credible, well-governed regulatory pathways for advanced therapies will attract patients, investment, and talent. Countries that maintain blanket prohibition without offering practical alternatives will see their patients travel elsewhere. The regulatory gap is not closing in the near term. It may, in fact, widen.
Conclusion
The legal difference between what a physician can offer in Bogota versus Boston is real, significant, and not adequately explained by saying that one country is safe and the other is not. The FDA’s restrictive framework reflects a specific historical and institutional logic that made sense for mass-market pharmaceutical drugs but fits poorly with personalized biological therapies. Colombia’s framework reflects a different logic, one centered on institutional standards, physician accountability, and risk stratification.
For patients who have exhausted conventional options or who are managing conditions where timing matters, the regulatory gap has direct personal consequences. Understanding it accurately, rather than through the lens of either uncritical promotion or reflexive dismissal, is the foundation for making an informed decision.
The right question is not whether a treatment is legal in the United States. The right question is whether it is being administered in a regulated, accountable clinical environment by qualified professionals using documented protocols. In Colombia, the answer to that question can be yes, and it can be verified.
If you are considering treatment and want to understand whether your specific condition and circumstances make you a good candidate, a consultation is the right starting point. The goal of that conversation is not to sell you a treatment. It is to give you an honest picture of what the evidence shows, what a protocol would involve, and what outcomes are realistic for someone in your situation.